Medical Devices Cybersecurity Requirements in the EU MDR and IVDR
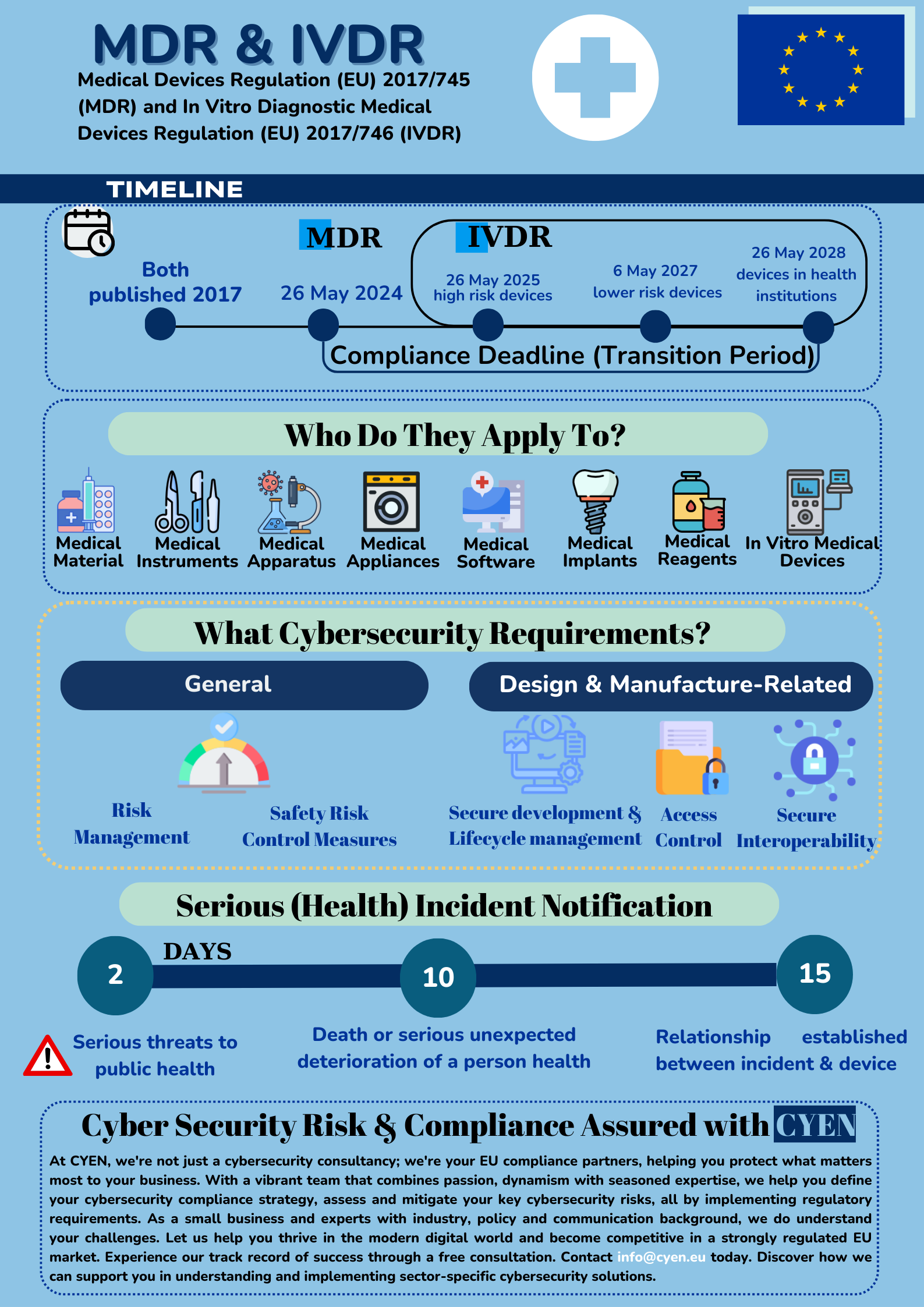
The EU Medical Devices Regulation 2017/745 & In Vitro Diagnostic Medical Devices Regulation 2017/746 are two EU regulations that introduce cybersecurity requirements for the medical devices made available on the EU market.
- Scope
MDR defines medical devices as diagnostic equipment, surgical instruments, monitoring devices, implantable devices, infusion pumps, and rehabilitation equipment. In vitro medical devices, as defined by the IVDR, are those medical devices used for in vitro diagnostics, e.g. devices which are used for the examination of specimens derived from the human body, such as blood or tissue samples, to provide information about a patient’s health condition.
- Novelties
Although MDR/IVDR main aim is not cybersecurity, new provisions introduce the notion of IT security. The security-related objectives of these Regulations are to achieve a high level of protection of the health of patients and users. The medical devices need to be properly examined, evaluated, monitored and supported throughout their life cycle.
- (Cyber)Security Requirements
Safety and effectiveness. The devices performance, safety and reliability should be as per the state of art. The devices need to be designed, manufactured and maintained throughout their life cycle in a way that prevents unauthorised access, protects against potential security threats, and safeguards the confidentiality, integrity, and availability of data (personal or non-personal). Manufacturers could achieve this by implementing secure software development practices, addressing vulnerabilities, and secure device software updates / patches.
Removal of IT-associated risk. The devices should be designed with robust risk management and risk control measures, in view to minimise the risk. Manufacturers will have to demonstrate they have assessed the appropriate risk and determined the proportionate security measures to be taken when manufacturing the device. Examples of appropriate risk mitigation measures include encryption, access controls, strong authentication or intrusion detection.
- Reporting of serious incidents
Manufacturers have to report to the relevant competent authorities serious incidents involving their devices on the EU market. Serious incident is any incident that directly or indirectly leading to:
- the death of a patient or other person
- the temporary or permanent serious deterioration of a patient’s or other person’s health
- a serious public health threat
To sum, all manufacturers of medical devices and in vitro diagnostics medical devices, or relevant vendors and providers who want to access or be placed on the EU market need to demonstrate specific cybersecurity measures are implemented and notify serious incidents.
NB: Other recent regulations, such as the EU NIS2 Directive places other cybersecurity / incident notification requirements for manufacturers (check CYEN’s analysis here: article, video, infographic). The new EU AI Act and the upcoming Cyber Resilience Act should be considered too. Check out our relevant videos and contact us if you like to understand whether or how to comply with the requirements applicable to your company or product.
EU MDR & IVDR Infographic
Recent Comments